Cardiac Arrhythmia
Our laboratory investigates the cellular and molecular mechanisms of arrhythmias and sudden cardiac death (SCD).
Our goal is to understand the emerging role of macromolecular ion channel complexes in the mechanisms of SCD in inherited diseases. Our recent research into the interactions between cardiac sodium and potassium channels provides an exceptional opportunity to define the molecular basis of SCD. We have found that cardiac excitability is controlled through interaction between NaV1.5, the main sodium channel in the heart ventricles, and the inward rectifier potassium channel Kir2.1. These ion channels form macromolecular complexes called channelosomes that physically interact with other partner proteins, including adapters, scaffolds, and enzymes that regulate the function of electrical currents. Both NaV1.5 and Kir2.1 are directly implicated in inherited diseases known as channelopathies. Andersen-Tawil syndrome type 1 (ATS1), also known as long QT syndrome type 7, is mediated by loss-of-function mutations in the gene KCNJ2, which encodes Kir2.1. Gain-of-function mutations in the same gene cause short QT syndrome type 3 (SQTS3). Loss-of-function defects in SCN5A, the gene encoding NaV1.5, give rise to Brugada syndrome (BrS). Kir2.1 and NaV1.5 function is also disrupted by defects in the dystrophin gene that underlie Duchenne muscular dystrophy (DMD), resulting in arrhythmias and SCD. The disturbed function of Kir2.1 and NaV1.5 in DMD exemplifies the importance of ion-channel interactions with multiple other proteins in the mechanisms of cardiac diseases.
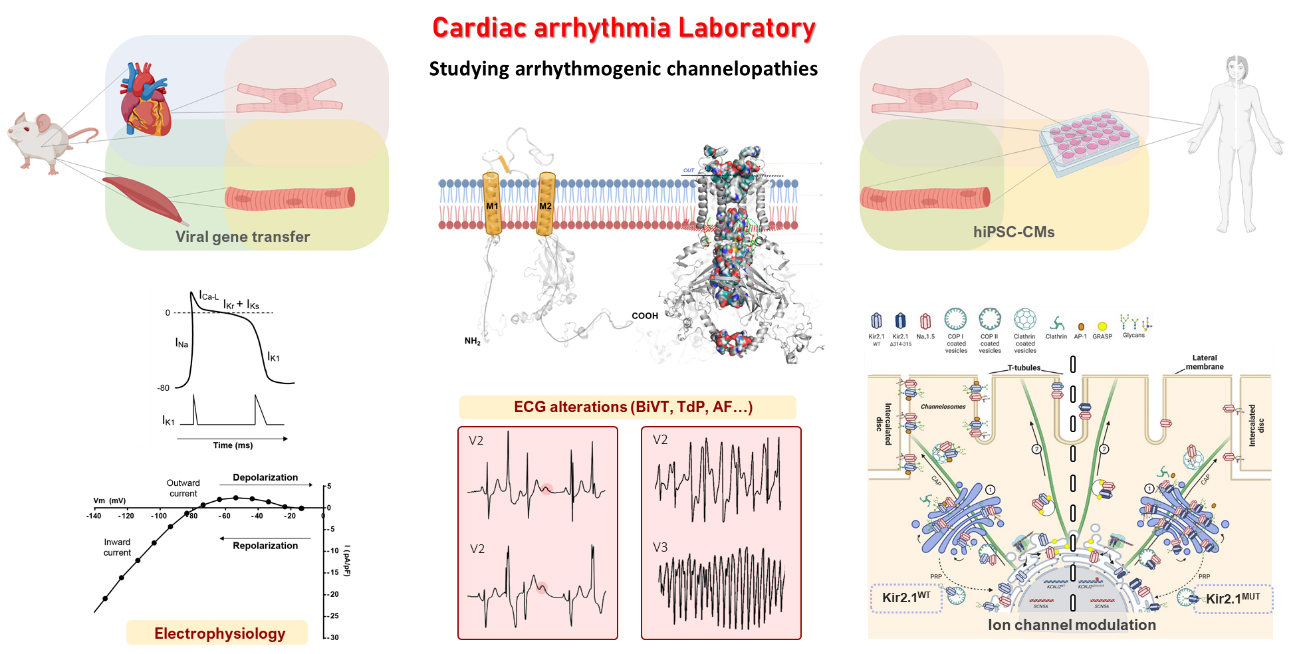
Our multidisciplinary approach includes the use of transgenic mouse models generated by adeno-associated virus (AAV)–mediated gene transfer and complementary experiments with human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs). The hiPSC-CMs are generated either from somatic cells (epithelial fibroblasts) of patients carrying a specific mutation or from non-mutated cells that are subsequently modified by CRISPR/Cas9 genomic editing. Our experiments with hiPSC-CMs are made possible through collaborations with three Spanish hospitals: Virgen de las Nieves Hospital in Granada, La Fe Hospital in Valencia, and Central University Hospital of Asturias in Oviedo.
The broad set of complementary techniques we use to investigate the electrophysiological mechanisms of inheritable arrhythmias includes electrocardiography (ECG), intracardiac stimulation, patch-clamping, optical mapping, RNA sequencing, western blotting, proteomics, and immunocytochemistry.
Our most recent findings include the exciting discovery that hiPSC-CMs derived from patients with DMD have a dysfunctional NaV1.5-Kir2.1 channalosome that can be rescued by expression of the scaffolding protein α1-syntrophin (published in eLife). Expression of α1-syntrophin (encoded by the SNTA1 gene) restored the localization of the ion channels in the hiPSC-CM membranes, and the subsequent increase in cell excitability prevented arrhythmias in hiPSC-CM monolayers. In addition, we have generated an AAV-mediated cardiac-specific mouse model of ATS1 that expresses a trafficking-deficient mutant Kir2.1 protein (Kir2.1∆314-315). These mice reproduce the cardiac electrical defects observed in patients with ATS1. The defects are caused by a dual dysfunction produced by the KCNJ2 mutation: one in the sarcolemma, which results in reduced excitability and abnormal conduction, and the other in the sarcoplasmic reticulum (SR), where the mutant Kir2.1 channels directly alter calcium dynamics across the SR (published in Nature Cardiovascular Research).
Our laboratory studies are complemented by computer modeling of ion channel structure and channel–ligand interactions, and more recently we have used computer models to design antiarrhythmic drugs in parallel with experimental testing in human and animal models. We expect our studies to yield important information with potential to improve antiarrhythmic therapy and help prevent SCD in patients suffering from inheritable cardiac diseases.



