Arritmias Cardíacas
El enfoque general del laboratorio del Dr. Jalife es la comprensión de los mecanismos celulares y moleculares de las arritmias y la muerte súbita cardíaca (MSC).
El objetivo del laboratorio es comprender el papel emergente de los complejos macromoleculares de canales iónicos en los mecanismos de la MSC en enfermedades hereditarias. Nuestra reciente identificación de la interacción del canal principal de sodio en los ventrículos (NaV1.5) con el canal rectificador de potasio (Kir2.1) en el control de la excitabilidad cardíaca, ofrece una oportunidad excepcional para definir las bases moleculares de la MSC. La evidencia indica que NaV1.5 y Kir2.1 forman "canalosomas" y que interactúan físicamente con proteínas afiliadas (adaptadores, andamios y enzimas reguladoras de la función de las corrientes eléctricas). Ambos canales tienen vínculos directos con enfermedades hereditarias que se conocen con el nombre de “canalopatías”. El Síndrome de Andersen-Tawil tipo 1 (del inglés, ATS1, englobado dentro del síndrome de QT largo tipo 7) está mediado por mutaciones de pérdida de función en el gen que codifica Kir2.1 (KCNJ2). Mutaciones de ganancia de función en el mismo gen (KCNJ2) causan el Síndrome de QT Corto tipo 3 (del inglés, SQTS3). Los defectos genéticos tráfico-deficientes en el gen que codifica a NaV1.5 (SCN5A) dan lugar al Síndrome de Brugada (BrS). Por otro lado, los defectos en el gen de la distrofina que resultan en la Distrofia Muscular de Duchenne (DMD) conducen a una disfunción de ambos canales y a MSC, lo que resalta aún más la relevancia de la interacción de los canales iónicos con otras proteínas en las enfermedades cardíacas.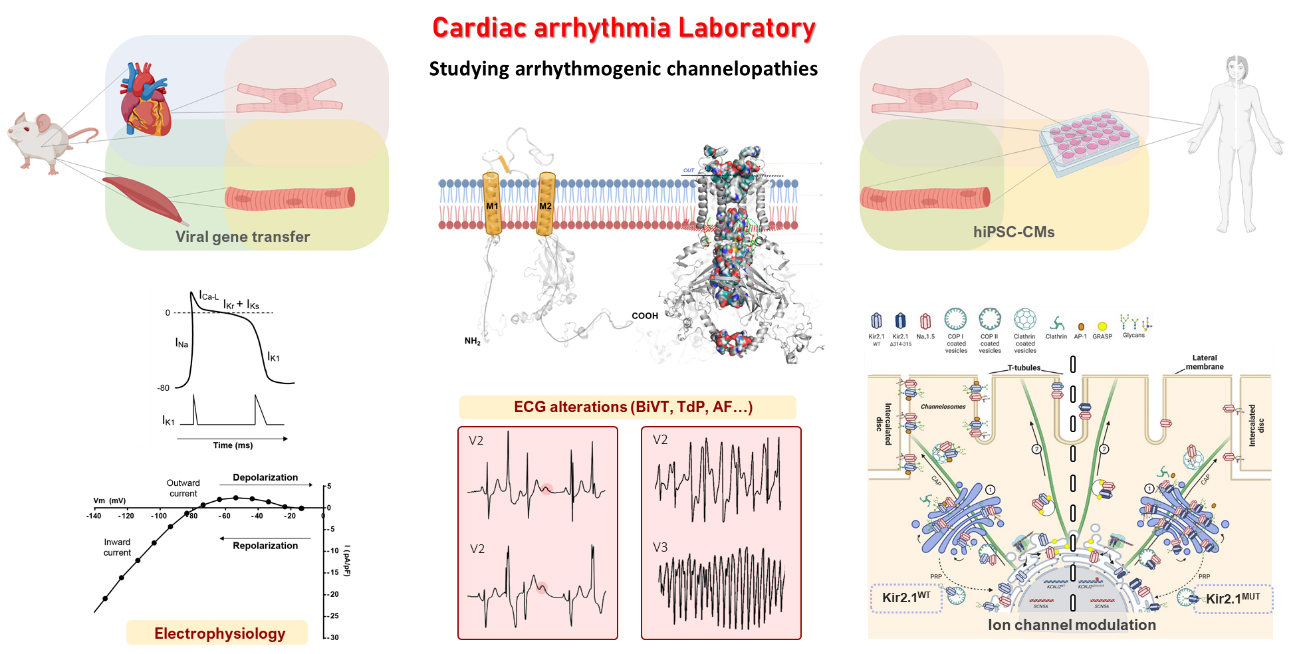
Dicho objetivo lo llevamos a cabo mediante un enfoque multidisciplinario, que engloba el uso de modelos animales de ratón generados mediante la transferencia de genes por virus adenoasociados, así como el uso de cardiomiocitos derivados de células madre humanas pluripotentes (del inglés, hiPSC-CMs), ya sea mediante la edición genómica CRISPR/Cas9 de células control o mediante su generación desde células somáticas (fibroblastos epiteliales) de pacientes y voluntarios sanos. Esta tecnología la podemos implantar gracias a la colaboración con el Hospital Virgen de las Nieves de Granada, el Hospital La Fe de Valencia y el Hospital Universitario Central de Asturias de Oviedo.
Entre nuestras principales técnicas para comprender los mecanismos electrofisiológicos de las enfermedades cardíacas que estudiamos, destaca el estudio del electrocardiograma (ECG), la estimulación intracardiaca, la fijación de voltaje en parches de membrana (‘patch-clamp’), el mapeo óptico, RNAseq, western blot, proteómica, e inmunohistoquímica. Nuestros estudios se complementan con el modelado computacional de la estructura del canal iónico, las interacciones canal-ligando y, más recientemente, el diseño de fármacos antiarrítmicos en paralelo con la experimentación en modelos humanos y animales.
Nuestros hallazgos más recientes incluyen que iPSC-CM de pacientes con distrofia muscular de Duchenne (DMD) tienen un canalosoma NaV1.5-Kir2.1 disfuncional que puede ser rescatado por la proteína de andamiaje sintrofina-α1 (gen SNTA1), restaurando los canales iónicos en la membrana (publicado en la revista eLife). En consecuencia, la excitabilidad celular se normaliza y se previenen las arritmias en las monocapas de cardiomiocitos derivados de pacientes con DMD. Además de esto, hemos generado un modelo de ratón específico cardíaco mediado por virus adenoasociado (AAV) del síndrome de Andersen-Tawil tipo 1 (ATS1) que expresa una proteína mutante deficiente en tráfico (Kir2.1∆314-315), que reproduce los defectos eléctricos observados en pacientes con ATS1. Dichos defectos son debidos a una disfunción dual producida por la mutación KCNJ2: una en el sarcolema, que resulta en excitabilidad reducida una conducción anormal, y la otra en el retículo sarcoplásmico, donde los canales mutantes SR Kir2.1 alteran directamente la dinámica del calcio (publicado en la revista Nature Cardiovascular Research).
De este modo, nos acercaremos al diseño de nuevas dianas terapéuticas y fármacos que puedan ser estudiados en los modelos humanos y animales generados con el objetivo de mejorar los tratamientos actuales y reducir la tasa de MSC en los pacientes que sufren estas y otras canalopatías.



